"Eu vou de órtese, porque eu quero ir representando os meninos de distrofia", disse o menino Pedrinho para sua mãe ao ser convidado para um desfile de moda infantil
Larissa Carvalho para opovo.com.br

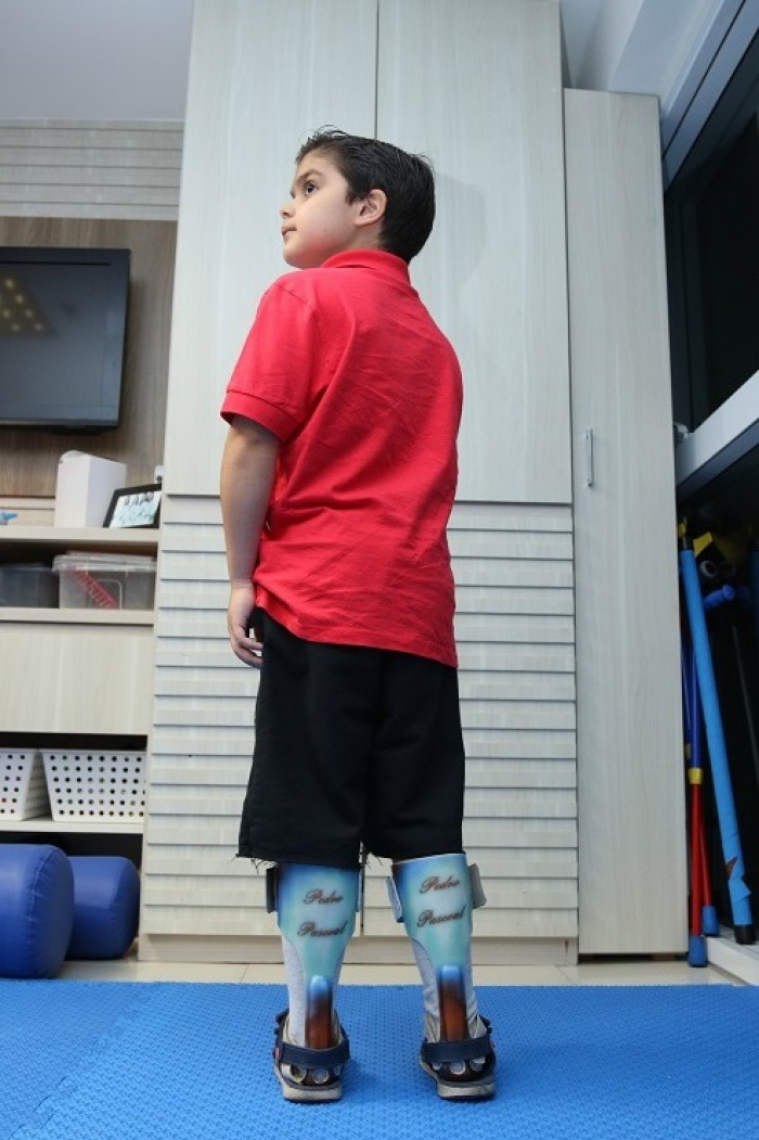
Pedro Pascoal, 10, tem distrofia muscular e se torna exemplo para outras crianças com a mesma doença, depois de participar de desfile no Shopping Iguatemi Fortaleza.(Foto: Tatiana Fortes/O POVO)
“Você vai de tênis?”, disse a mãe para o filho que havia sido convidado para um desfile de moda infantil. “Ele disse: 'Não, eu vou de órtese, porque eu quero ir representando os meninos de distrofia'”. Na tarde do último domingo, 7, o menino Pedro Pascoal, 10 anos, participou do desfile Estilinho, no Shopping Iguatemi Fortaleza. Pedrinho, como é chamado, tem Distrofia Muscular de Duchenne (DMD), um dos mais de 30 tipos de doenças genéticas relacionadas ao músculo.
“Filho, como você vai fazer? Você vai ver a multidão lá [no desfile]. Ele disse: 'Não tem problema. Eu vou colocar um óculos escuro e aí eu não vou ficar mais com vergonha'”, disse a mãe, Ozilene Pascoal, 43. Pedrinho é uma criança bem tímida, como descreve sua mãe. Mas isso não o impediu de participar do desfile, junto de suas órteses para representar as outras crianças, que têm algum tipo de deficiência.
O menino foi diagnosticado com a condição quando tinha apenas quatro anos de idade. “O Pedrinho era muito querido e quando eu tive ele, ele chorava muito, muito mesmo. Eu levava ele aos médicos e diziam que ele não tinha nada”, relata. Após várias idas aos médicos, o menino fez exames com o médico neurologista, quando foi descoberto. Um dos sintomas desse tipo de disfunção são quedas frequentes ao andar, impossibilidade de correr, fortes dores, além das panturrilhas aumentadas.
“Eles não conseguem correr, por conta da fraqueza muscular. Então, as outras crianças acabam excluindo e eles se sentem só”, conta a mãe de Pedrinho. “Esses meninos são excluídos da sociedade, eles não participam desses eventos”. Conforme estimativas da Organização Mundial da Saúde (OMS), existem mais de oito mil doenças raras catalogadas. 6% a 8% da população tem algum tipo dessas doenças.
Confira o vídeo do momento:
 “O que leva o progresso da doença é a sobrecarga de atividade física. Ele pode fazer, só que a atividade tem que chegar até o ponto dele não ter fadiga”, diz a fisioterapeuta. O menino ainda faz visitas frequentes ao cardiologista, neurologia, nutricionista, pediatra, pneumologista, psicólogo, além de hidroterapia. Recentemente, Pedrinho passou por uma cirurgia devido ao encurtamento da panturrilha.
“O que leva o progresso da doença é a sobrecarga de atividade física. Ele pode fazer, só que a atividade tem que chegar até o ponto dele não ter fadiga”, diz a fisioterapeuta. O menino ainda faz visitas frequentes ao cardiologista, neurologia, nutricionista, pediatra, pneumologista, psicólogo, além de hidroterapia. Recentemente, Pedrinho passou por uma cirurgia devido ao encurtamento da panturrilha.
Segundo a mãe, o convite para o desfile surgiu quando eles estavam passeando no shopping e Pedrinho pediu para entrar em uma loja, pois havia gostado de uma roupa. “A moça abordou ele e disse: 'Você é tão lindo! Você não quer desfilar?'”. Ozilene levou o menino para experimentar a roupa escolhida por ele mesmo. “Ele foi e para a minha surpresa, ele disse que ia desfilar”, conta.
“Eu achei muito lindo isso dele, porque partiu dele. Eu fiquei muito emocionada porque ele sente na pele o que é ser excluído. Ele sente a dor de não ter amigos, de não chamarem ele para brincar. Ele se sente diferente das pessoas, mas ele quer ser aceito”.
Diagnóstico e tratamento
Além da escola, a rotina de Pedrinho inclui sessões de fisioterapia motora com a fisioterapeuta e especialista em Neurologia infantil, Nacha Cunha, durante todos os dias da semana, na Clínica Superare. “A criança nasce normal, com desenvolvimento neuropsicomotor normal. E por volta de cinco a seis anos, a criança começa a cair bastante. Os pais procuram o ortopedista e o neurologista para o diagnóstico", explica sobre os primeiros momentos.
“O que leva o progresso da doença é a sobrecarga de atividade física. Ele pode fazer, só que a atividade tem que chegar até o ponto dele não ter fadiga”, diz a fisioterapeuta. O menino ainda faz visitas frequentes ao cardiologista, neurologia, nutricionista, pediatra, pneumologista, psicólogo, além de hidroterapia. Recentemente, Pedrinho passou por uma cirurgia devido ao encurtamento da panturrilha.
Os pais de Pedrinho, Ozilene Pascoal, 43, e Jean de Carvalho, 43, fazem parte da Associação Cearense de Distrofias Musculares (ACDM). A instituição é formada por pais, pacientes e amigos no objetivo de orientar e conscientizar sobre a convivência com as distrofias musculares. A ACDM assiste 20 famílias de forma direta e é composta por cerca de 60 pessoas.
Associação Cearense de Distrofias Musculares (ACDM) - Rua das Carnaúbas, 167, no bairro Itaperi (próximo ao Zoológico Municipal Sargento Prata)


















